- HÉMOPHILIE
- HÉMOPHILIEL’hémophilie est une affection hémorragique héréditaire due à la diminution ou à l’absence d’un facteur de coagulation dit facteur antihémophilique. L’existence de facteurs antihémophiliques A et B permet de distinguer deux formes différentes d’hémophilies: l’hémophilie A et l’hémophilie B, la première étant la plus fréquente. Bien que rare, cette affection pose de nombreux problèmes du fait de la gravité du syndrome hémorragique. Certes, aujourd’hui l’hémophilie met rarement la vie en danger, mais la localisation des hémorragies et, en particulier, les hémarthroses risquent trop souvent de transformer le malade en infirme et en inadapté social.La description, autour des années 1970, de formes modérées de l’hémophilie a posé le problème des méthodes biologiques sensibles nécessaires pour permettre leur diagnostic. Dans le même temps, d’importants progrès thérapeutiques ont été réalisés par la préparation de fractions plasmatiques concentrées contenant l’un ou l’autre des facteurs antihémophiliques, ce qui permet de corriger le trouble de la coagulation par le biais d’un traitement substitutif. Malheureusement, cette thérapeutique transfusionnelle a été à l’origine de la contamination, dans les années 1980, d’un grand nombre d’hémophiles par des virus, ceux des hépatites (B et C) et celui du déficit immunitaire humain (VIH).Une utilisation plus précoce des produits chauffés aurait permis de limiter la contamination par le virus VIH. À la suite de ce drame, des progrès considérables ont été réalisés dans la purification et la réduction du risque viral des produits; aujourd’hui, les produits sont de «très haute pureté» ou «ultrapurs», à risque viral nul vis-à-vis du virus VIH et des virus des hépatites B et C.Histoire de l’hémophilieL’hémophilie n’a été acceptée comme entité clinique qu’à partir du XIXe siècle. Cependant, cette diathèse hémorragique congénitale et familiale était déjà connue depuis longtemps puisque, dès le IIe siècle avant J.-C., le Talmud de Babylone dispensait du rite de la circoncision le troisième d’une fratrie dont les deux frères aînés étaient morts d’hémorragies après circoncision.La première description de cette affection dans la littérature médicale est due à J. C. Otto (1803). L’auteur évoquait déjà la transmission génétique, qui fut précisée par C. F. Nasse en 1820, et le nom d’hémophilie fut attribué à la maladie par F. Hopff en 1828. Toutefois, ce n’est réellement qu’en 1911 que W. Bulloch et P. Fildes, revoyant les données, souvent confuses, accumulées sur l’hémophilie, en définissent les principaux critères: tendance au saignement excessif depuis l’enfance, existence d’autres cas identiques dans une même famille; transmission par des femmes apparemment normales, et allongement du temps de coagulation. L’insuffisance des connaissances sur le mécanisme de l’hémostase a, pendant longtemps, conduit à confondre sous le nom d’«hémophilie» toute diathèse hémorragique caractérisée par un trouble de la coagulation in vitro.Les étapes ultérieures dans l’individualisation de l’hémophilie sont parallèles au progrès de la physiologie de la coagulation. En 1937, A. J. Patek et F. H. Taylor proposent de nommer «globuline antihémophilique» le facteur de coagulation absent ou diminué dans le plasma des hémophiles. Entre 1947 et 1953, différents auteurs (A. Pavlosky, I. Schulman et C. H. Smith, J. C. F. Poole) montrèrent qu’il existait une correction mutuelle entre le sang de deux hémophiles différents; ils postulèrent de ce fait l’existence de deux types d’hémophilies difficiles à classer, car leurs manifestations cliniques et leur transmission génétique sont identiques dans les deux cas. La mise en évidence et l’étude des caractères de deux protéines plasmatiques permirent en 1954 au Comité international de nomenclature de définir les deux groupes d’hémophilies: hémophilie A liée à un déficit en facteur VIII (facteur antihémophilique A) et hémophilie B due à un déficit en facteur IX (facteur antihémophilique B).Parallèlement, la transmission héréditaire de la maladie fut éclaircie grâce à l’étude des généalogies des familles d’hémophiles et grâce à la découverte d’une affection identique chez le chien, qui a permis de réaliser des croisements multiples.Le clonage du gène du facteur IX par Choo et ses collaborateurs en 1982 et celui du facteur VIII par Gitschier et ses collaborateurs en 1984 ont permis, d’une part, la détection des conductrices et le diagnostic anténatal par analyse génotypique de l’ADN du chromosome X et, d’autre part, la synthèse du facteur VIII par génie génétique (facteur VIII recombinant).GénétiqueL’hémophilie se transmet selon un mode récessif lié au sexe, c’est-à-dire que, le gène anormal étant situé sur le chromosome X-, elle ne s’exprime que chez les sujets de sexe masculin, alors que les femmes «conductrices» transmettent la maladie mais sont cliniquement indemnes. Les études généalogiques ont fait apparaître, par exemple, dans la descendance de la reine Victoria d’Angleterre que la tare peut se transmettre par les femmes pendant plusieurs générations. L’hémophilie féminine reste exceptionnelle; une femme hémophile, homozygote pour la tare, proviendrait en effet de l’union, très rare, statistiquement parlant, d’un homme hémophile avec une femme conductrice.L’hémophilie peut apparaître sporadique, soit parce que l’ignorance des antécédents familiaux ne permet pas de la déceler, parfois du fait du petit nombre de descendants mâles dans une famille, soit parce qu’il y a une néomutation au niveau des gamètes des grands-parents maternels ou de la mère de l’enfant hémophile. Les néomutations sont évaluées à 30 p. 100 des cas environ.L’apport des méthodes de biologie moléculaire a été considérable, permettant l’analyse des défauts géniques – responsables de l’hémophilie A et de l’hémophilie B – et la détection des conductrices. Des mutations ou des délétions ont été identifiées au niveau des gènes du facteur VIII et du facteur IX par amplification génique, électrophorèse en gradient de gel dénaturant et séquençage. Le gène du facteur VIII est situé dans la partie la plus terminale du bras long du chromosome X et comprend 186 kilobases; l’ARNm mesure 9 kilobases et code pour une protéine de 2 351 acides aminés. Le gène du facteur IX est situé dans la région distale du bras long du chromosome X et s’étend sur 34 kilobases; la partie codante est constituée de 1 248 paires de bases codant pour 415 acides aminés.Symptomatologie cliniqueLa symptomatologie clinique se résume aux manifestations hémorragiques et à leurs séquelles. Les premières hémorragies surviennent rarement durant la période néo-natale, sauf parfois après circoncision. Dans les formes graves, c’est habituellement vers la fin de la première année, et surtout lors des premiers pas, que l’apparition d’ecchymoses et d’hématomes conduit au diagnostic. En revanche, dans les formes modérées, l’hémophilie peut n’être reconnue que vers l’adolescence, et parfois à l’occasion d’une intervention chirurgicale.L’hémophilie se manifeste par des hémorragies apparemment spontanées ou provoquées par des traumatismes même minimes; ces hémorragies peuvent être extériorisées ou se produire à l’intérieur des tissus.Hémorragies extérioriséesCutanées ou muqueuses, les hémorragies extériorisées peuvent, par leur abondance et leur répétition, entraîner une anémie aiguë, autrefois mortelle. Elles surviennent au niveau de plaies cutanées, même légères, de lésions buccales: morsures de la langue, plaie du frein de la langue (elles sont alors particulièrement difficiles à contrôler), de la muqueuse nasale, du tractus digestif ou rénal. Les hémorragies postopératoires sont la règle en l’absence d’un traitement substitutif efficace.Hémorragies intratissulairesL’hémorragie peut se produire dans le tissu cellulaire (hématome sous-cutané) et dans les muscles (hématomes intramusculaires). Ces hématomes intramusculaires, de localisation variée, entraînent par leur importance des troubles de compression vasculaire ou nerveuse et conduisent parfois à l’évacuation chirurgicale pour pallier les séquelles fonctionnelles.Une localisation très particulière est constituée par les hémarthroses (hémorragies intra-articulaires). Rares dans les formes modérées, elles apparaissent régulièrement dans les formes graves; elles se situent principalement au niveau des genoux, des chevilles et des coudes, atteignant plus rarement les hanches et les poignets. Après une phase aiguë, avec douleurs et signes inflammatoires, elles peuvent devenir chroniques, surtout en raison de récidives fréquentes au niveau d’une même articulation. Il apparaît alors une déformation variable de l’articulation, accompagnée souvent d’une atrophie musculaire et, surtout, de lésions cartilagineuses et osseuses plus ou moins importantes qui témoignent de l’existence d’hémorragies sous-périostées ou intra-osseuses. Il s’agit d’une véritable arthropathie avec évolution vers l’ankylose fibreuse. La localisation des arthropathies et leur importance font tout le pronostic fonctionnel de l’hémophilie. Les autres localisations hémorragiques (hémorragies cérébrales en particulier) sont plus rares mais ne sont pas exceptionnelles.L’intensité du sydrome hémorragique et la fréquence des accidents sont fonction du degré de l’hémophilie, mais constantes chez un même malade. Dans les formes graves de la maladie, on observe l’association d’ecchymoses fréquentes, d’hématomes étendus, d’hémarthroses répétées, le contexte clinique s’accompagnant de désordres biologiques importants. Dans les formes modérées, les hémorragies provoquées restent les manifestations les plus habituelles de la maladie. Enfin, certaines formes atténuées peuvent passer inaperçues jusqu’à une période tardive de l’existence, ou ne se révèlent qu’à l’occasion d’une intervention chirurgicale ou d’un traumatisme important.Diagnostic biologiqueL’anomalie fondamentale de la coagulation dans l’hémophilie est l’absence ou la diminution du facteur antihémophilique A ou B (facteur VIII ou IX); c’est cette absence ou cette diminution qu’il faut savoir mettre en évidence. Classiquement, l’hémophilie se traduit en effet par un allongement du temps de coagulation du sang total. En réalité, cela ne s’observe que dans les formes graves de l’affection (environ 50 p. 100 des cas) où le déficit en facteur antihémophilique est absolu. Mais dans les formes modérées, dès que le taux de facteur antihémophilique atteint 3 à 6 p. 100 de la normale, le diagnostic d’hémophilie ne peut être écarté avant qu’on ait eu recours à des méthodes plus sensibles que celle du temps de coagulation, qui est ici normal. Le temps de céphaline activé, ou temps de coagulation du plasma en présence d’un excès de phospholipide (remplaçant le facteur plaquettaire 3), apparaît actuellement comme la méthode la plus sensible et la plus simple pour rechercher l’existence d’un trouble de la coagulation. Il doit nécessairement être suivi de la mesure spécifique des facteurs VIII et IX, mesure indispensable, d’une part, pour préciser le type de l’hémophilie en cause et, d’autre part, pour apprécier le degré de la déficience. On définira ainsi: des formes graves , où le taux de facteur VIII ou IX est inférieur à 1 p. 100; des formes modérées , où il varie entre 1 et 5 p. 100; des formes atténuées , où il est compris entre 5 et 30 p. 100.PathogénieLe trouble de la coagulation dans l’hémophilie est lié à l’absence d’activité biologique des facteurs antihémophiliques A et B intervenant tous deux à une phase initiale de la coagulation, la thromboplastinoformation endogène [cf. HÉMOSTASE ET HÉMORRAGIES]. Cette absence d’activité a été considérée habituellement comme l’effet d’un déficit lié à l’absence de synthèse des facteurs VIII ou IX. Des travaux récents, utilisant des méthodes immunologiques, ont montré en fait la distinction entre deux groupes d’hémophilie: l’hémophilie dite A- ou B- se définit par l’absence d’activité biologique ou antigénique du facteur antihémophilique en cause; l’hémophilie dite A+ ou B+, beaucoup plus rare (10 p. 100 des cas), se caractérise par la présence dans le plasma du malade d’une molécule biologiquement inactive, mais possédant une activité antigénique identique à celle du facteur normal (VIII ou IX). Il est actuellement possible de doser les antigènes spécifiques du facteur VIII (VIII: CAg).TraitementIl n’existe actuellement aucun traitement curatif de l’hémophilie. Toute thérapeutique est donc essentiellement substitutive et consiste à augmenter le taux de facteur VIII ou IX à un niveau suffisant pour assurer une hémostase normale par l’apport de fractions plasmatiques concentrées de facteur VIII ou de facteur IX.Dans l’hémophilie A , des concentrés plasmatiques ou de facteur VIII recombinant sont actuellement disponibles. Les centres français de fractionnement du plasma fabriquent, depuis 1988, des concentrés de facteur VIII de très haute pureté. La purification du facteur VIII se fait par chromatographie, ce qui permet une concentration du facteur VIII d’environ 10 000 fois par rapport au plasma. Ces concentrés subissent un procédé de réduction virale par solvant-détergent qui inactive les virus à enveloppe lipidique (virus de l’immunodéficience humaine, virus des hépatites B et C, etc.) mais non les virus dits «nus» ou «non enveloppés» (hépatite A, parvovirus, etc.).Des concentrés plasmatiques de facteur VIII obtenus par immunopurification et produits aux États-Unis sont actuellement disponibles: l’Hemofil (Baxter-Hyland) et le Monoclate pasteurisé (Rhone-Poulenc/Armour/Rorer). La purification du facteur VIII est réalisée par immunopurification à l’aide d’anticorps monoclonaux de souris antifacteur VIII pour l’Hemofil M ou antifacteur Willebrand (protéine porteuse du facteur VIII dans le plasma) pour le Monoclate pasteurisé, ce qui permet d’obtenir un facteur VIII ultrapur: l’adjonction d’albumine humaine est cependant nécessaire à la stabilisation du facteur VIII dans le produit final. Le procédé de réduction virale et le solvant-détergent pour l’Hemofil M et une pasteurisation pour le Monoclate pasteurisé (procédé qui inactive les virus enveloppés et certains virus nus).Deux concentrés de facteur VIII recombinant, Recombinate (Baxter-Hyland) et Kogenate (Bayer-Miles), sont fabriqués aux États-Unis. Leur production a l’avantage d’être complètement indépendante du plasma. Les cellules qui fabriquent le facteur VIII sont des cellules d’ovaire (Recombinate) ou de rein (Kogenate) de hamster chinois. Le surnageant de culture subit une immunopurification par des anticorps monoclonaux de souris antifacteur VIII; l’adjonction d’albumine humaine pasteurisée est nécessaire à la stabilisation du facteur VIII dans le produit final. Le Recombinate a obtenu l’autorisation de mise sur le marché en Europe en mai 1993; le Kogenate sera disponible dès qu’il aura obtenu son autorisation de commercialisation en Europe.L’efficacité clinique et biologique de ces différents concentrés est comparable. Aucune donnée scientifique ne permet de faire à l’heure actuelle un choix clair et définitif entre ces différents concentrés. L’ultrapurification réduit le risque viral résiduel ainsi que celui d’altération de la fonction immune qui a été décrite avec des concentrés de pureté intermédiaire. S’il existe un déficit immunitaire, il paraît souhaitable d’utiliser un concentré de facteur VIII immunopurifié ou recombinant. La surveillance des patients traités avec ces nouveaux concentrés doit être étroite et régulière du fait des risques potentiels liés à la présence d’anticorps de souris, de traces d’ADN et de protéines de hamster, etc. Enfin, les études scientifiques comparatives indiqueront si le risque d’apparition d’anticoagulant circulant augmente avec la purification des produits.Dans l’hémophilie A atténuée , il existe une alternative au traitement transfusionnel: la déamino-D-vasopressine commercialisée en France sous le nom de Minirin, qui relargue le facteur VIII dans le sang circulant. Ce traitement n’est pas efficace chez tous les patients.Dans l’hémophilie B , les concentrés de facteur IX de très haute pureté préparés par les centres de fractionnement ont remplacé, depuis 1988, la fraction PPSB. Ces concentrés sont soumis à un traitement d’inactivation virale par solvant-détergent. Il existe aussi un concentré de facteur IX immunopurifié (Mononine de la firme Rhone-Poulenc/Armour/Rorer) en cours d’évaluation et qui pourrait, du fait d’une purification plus poussée, avoir une indication chez les hémophiles B avec déficit immunitaire.Le rythme des injections quotidiennes de concentrés de facteur VIII ou de facteur IX est fonction de la durée de vie du facteur antihémophilique en cause. La demi-vie du facteur VIII est de dix à douze heures, celle du facteur IX, de dix-huit à vingt-quatre heures. La dose utile est donnée par un calcul simple tenant compte du volume plasmatique du malade et du taux que l’on désire obtenir in vivo: de 10 à 40 p. 100 en fonction de l’importance de l’accident hémorragique. Chaque unité (U) de facteur VIII injectée par kilogramme de poids augmente le taux de facteur VIII circulant de 2 p. 100, chaque unité de fateur IX augmente le taux de facteur IX circulant de 1,5 p. 100. environ Une dose initiale de 20 U/kg élève le taux de facteur VIII à 40 p. 100 et suffit pour arrêter un saignement. En chirurgie, une dose de 50 U/kg est nécessaire suivie de deux ou trois injections par jour d’environ 20 à 30 U/kg jusqu’à cicatrisation complète.Le traitement de l’hémophilie est habituellement palliatif Il peut être prophylactique (20 U/kg toutes les quarante-huit heures) et administré pendant des périodes de durée variable dans certaines circonstances (hémarthroses récidivantes, stress, vacances, activité professionnelle, etc.). Les tentatives du traitement prophylactique continu, qui paraîtrait justifié dans cette affection éliminant virtuellement les hémorragies spontanées et le développement de l’atteinte articulaire, ont été limitées par le risque viral. En dehors des complications infectieuses, le traitement transfusionnel risque de déterminer chez le receveur la formation d’anticorps spécifiques que l’on appelle anticoagulants circulants. De 10 à 15 p. 100 des hémophiles A majeurs et également un petit nombre d’hémophiles modérés s’immunisent contre le facteur VIII. Le risque est beaucoup plus faible dans l’hémophilie B. L’apparition d’un anticoagulant circulant complique considérablement le traitement substitutif, qui devient inefficace si le titre de l’anticorps est élevé. Les moyens thérapeutiques sont alors les suivants: induction d’une tolérance immune par stimulation antigénique continue avec le facteur VIII ou le facteur IX, injections de fractions animales (facteur VIII porcin dans l’hémophilie A en l’absence d’immunité croisée), de facteurs du complexe prothrombique activés (Autoplex, Feiba), de facteur VII activé (Acset).Dans certains types d’hémorragies, un traitement local associé peut être nécessaire: compression (éponge synthétique avec ou sans agents coagulants tels que la thrombine), utilisation de colle biologique, application de gouttières en résine acrylique dans le cas d’extraction dentaire par exemple, de gouttières plâtrées dans le cas des hémarthroses, etc. L’importance de la rééducation musculaire chez les hémophiles, après immobilisation prolongée pour hémarthroses ou hématomes musculaires, ne doit pas être méconnue. C’est dire la nécessité d’une coopération permanente entre l’hématologiste, le stomatologiste, l’orthopédiste et le rhumatologue en vue d’une surveillance régulière du malade, afin de lui permettre d’atteindre l’âge adulte avec un minimum de séquelles fonctionnelles.La guérison des hémophiles n’est pas utopique. Elle a déjà été réalisée par la greffe de tissu producteur de facteur VIII (greffe hépatique). La thérapie génique n’en est qu’à ses balbutiements, mais elle est potentiellement l’une des plus importantes applications des recherches récentes à la médecine.Diagnostic anténatalLa détection des conductrices d’hémophilie A ou B est, bien sûr, nécessaire pour le diagnostic anténatal. Autrefois fondée sur l’étude du phénotype, c’est-à-dire la comparaison des taux de facteur VIII (VIII: C ou VIII: CAg) et de facteur Willebrand pour l’hémophilie A, et sur celle des taux de l’activité et de l’antigène facteur IX pour l’hémophilie B, elle s’accompagne maintenant de l’étude du génotype fondée sur la mise en évidence de polymorphismes de restriction intra ou extragéniques utilisés comme marqueurs.Cette détection est suivie par un conseil génétique. Le diagnostic anténatal est réalisé depuis 1983 en France chez le fœtus de sexe masculin à partir de la dix-huitième semaine de grossesse; un prélèvement de sang fœtal pur au niveau du cordon, sous contrôle échographique, donne un diagnostic parfaitement fiable. Depuis 1985, l’utilisation des sondes permettant d’étudier les polymorphismes intragéniques autorise un dépistage plus précoce par étude de l’ADN du chromosome X fœtal à partir d’un prélèvement de villosité choriale à la dixième semaine. Cette technique n’est applicable que lorsque l’étude génotypique familiale est informative.Aspect psychologique et socialLa fréquence de l’hémophilie est souvent difficile à préciser, surtout pour ce qui est des formes modérées reconnues récemment. On admet actuellement qu’elle serait de 1 p. 10 000 habitants, l’hémophilie A représentant 85 p. 100 des cas.Bien que rare, cette maladie pose des problèmes psychologiques et sociaux. En effet, l’hémophile, atteint d’une affection chronique, avec le risque permanent d’une hémorragie, physiquement amoindri par les séquelles des hémarthroses, soumis à de nombreuses hospitalisations et à de longues périodes d’immobilisation, se heurte à des problèmes psychologiques et sociaux permanents. Une partie de ces problèmes a été résolue par la mise en route de l’autotraitement réalisé à domicile.L’enfant hémophile connaît des contraintes: restrictions imposées à son activité (en particulier dans le domaine des sports), absentéisme scolaire, anxiété du saignement, souvent aggravées par une protection de la mère et les conflits conjugaux fréquents dans les familles d’hémophiles. Tous ces éléments conduisent trop souvent à des modifications psychologiques importantes. À l’âge adulte, les problèmes professionnels dominent souvent, liés une fois encore à l’absentéisme, à une formation insuffisante et au mauvais choix du métier. La création d’une Association française des hémophiles permet de pallier certains de ces obstacles, grâce à la fois à son rôle d’information et de soutien. Elle contribue activement au développement de l’organisation des soins.La reconnaissance de centres de traitement de l’hémophilie regroupant spécialistes, hématologistes, orthopédistes, kinésithérapeutes, mais aussi psychologues et assistantes sociales a représenté une étape importante. Ce type de prise en charge, associé à la thérapeutique transfusionnelle, avait transformé la vie des hémophiles. Il ne faut pas que l’infection par le virus VIH détruise cet acquis, remettant en question le traitement et la confiance en l’équipe médicale. Plus que jamais l’hémophile doit pouvoir disposer au sein d’une structure spécialisée d’un diagnostic rapide et précis, d’un traitement efficace et sûr et des dispositions nécessaires pour une prise en charge de vingt-quatre heures sur vingt-quatre, d’un suivi régulier, des conseils d’experts et d’un bon niveau de communication.
hémophilie [ emɔfili ] n. f.• 1855; de hémo- et -philie♦ Génét., méd. Maladie héréditaire transmise par les femmes et qui se manifeste chez les individus mâles, due à la modification d'un gène porté par un chromosome sexuel, et se traduisant par une incapacité du sang à coaguler.
● hémophilie nom féminin Maladie héréditaire, récessive et liée au sexe (transmise par les femmes et n'atteignant que les hommes), caractérisée par une tendance plus ou moins grave aux hémorragies, du fait de l'insuffisance d'un facteur de coagulation A ou B dans le plasma.hémophilien. f. MED Maladie héréditaire, transmise par les femmes mais n'atteignant que les hommes, due à l'absence de certains facteurs plasmatiques de la coagulation et caractérisée par une tendance aux hémorragies répétées et abondantes.⇒HÉMOPHILIE, subst. fém.PATHOL. Affection congénitale et héréditaire caractérisée par un retard de la coagulation et des hémorragies graves apparaissant au moindre traumatisme. Hémophilie constitutionnelle, sporadique; arthropathie de l'hémophilie. La faiblesse d'esprit, la folie, l'hémophilie, la surdi-mutité, comme on le sait, sont des vices héréditaires (CARREL, L'Homme, 1935, p. 305).REM. 1. Hémophile, adj. et subst. (Malade) qui est atteint d'hémophilie (cf. Méd. Biol. t. 2, 1972). 2. Hémophilique, adj. Qui se rapporte à l'hémophilie. Arthrite hémophilique, arthropathies hémophiliques (RAVAULT, VIGNON, Rhumatol., 1956, p. 564).Prononc. et Orth. : [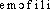 ]. Att. ds Ac. 1935. Étymol. et Hist. 1858 pathol. (NYSTEN, Dict. [11e éd., Baillière] ds Fr. mod. t. 39, p. 155). Composé de l'élém. hémo- (héma-) et de l'élém. -philie (-phile et -ie). Bbg. QUEM. DDL t. 5; t. 8 (s.v. hémophile, hémophilique).hémophilie [emɔfili] n. f.❖1 Didact. Disposition pathologique aux hémorragies prolongées, dues à l'absence d'un facteur de coagulation dans le sang, affection héréditaire transmise par les femmes uniquement aux enfants de sexe masculin.0 L'hémophilie est caractérisée par un grand retard de la coagulation sanguine avec diminution de la coagulabilité, tous les autres éléments physiques, chimiques et biologiques du sang, ainsi que l'endothélium vasculaire, étant normaux.M. Garnier et V. Delamare, Dict. termes techniques de médecine, art. Hémophilie.2 Cour. État pathologique, non héréditaire, qui offre certains caractères de l'hémophilie vraie, et qui peut affecter les deux sexes. || Hémophilie familiale, héréditaire.❖DÉR. Hémophile, hémophilique.
]. Att. ds Ac. 1935. Étymol. et Hist. 1858 pathol. (NYSTEN, Dict. [11e éd., Baillière] ds Fr. mod. t. 39, p. 155). Composé de l'élém. hémo- (héma-) et de l'élém. -philie (-phile et -ie). Bbg. QUEM. DDL t. 5; t. 8 (s.v. hémophile, hémophilique).hémophilie [emɔfili] n. f.❖1 Didact. Disposition pathologique aux hémorragies prolongées, dues à l'absence d'un facteur de coagulation dans le sang, affection héréditaire transmise par les femmes uniquement aux enfants de sexe masculin.0 L'hémophilie est caractérisée par un grand retard de la coagulation sanguine avec diminution de la coagulabilité, tous les autres éléments physiques, chimiques et biologiques du sang, ainsi que l'endothélium vasculaire, étant normaux.M. Garnier et V. Delamare, Dict. termes techniques de médecine, art. Hémophilie.2 Cour. État pathologique, non héréditaire, qui offre certains caractères de l'hémophilie vraie, et qui peut affecter les deux sexes. || Hémophilie familiale, héréditaire.❖DÉR. Hémophile, hémophilique.
Encyclopédie Universelle. 2012.